

400-820-8531




肝脏是机体核心代谢器官,其功能异常易引发肝硬化甚至肝细胞癌,脂质稳态紊乱是关键驱动因素。ATG9A是一种自噬相关跨膜蛋白及脂质翻转酶,参与调节脂质动态,但在肝脏代谢作用尚不明确。
2025年12月4日,电子科技大学张琳团队在Autophagy期刊上发表了题为“ATG9A-PLA2G6 axis reprograms phospholipid metabolism to drive metabolic liver disease and hepatocellular carcinoma”的研究文章,揭示 ATG9A 与 PLA2G6 结合形成调控轴,通过重编程磷脂代谢引发线粒体功能障碍,以非自噬依赖方式加剧肝脏炎症与纤维化、促进肝细胞癌进展,为代谢性肝病及肝细胞癌的治疗提供潜在新靶点。拜谱生物为该研究提供了DIA定量蛋白组和靶向脂质代谢组学技术支持。

英文标题:ATG9A-PLA2G6 axis reprograms phospholipid metabolism to drive metabolic liver disease and hepatocellular carcinoma(Autophagy IF 14.3)
中文标题:ATG9A-PLA2G6 轴通过重编程磷脂代谢来驱动代谢性肝病和肝细胞癌
客户单位:电子科技大学
研究材料:肝脏
拜谱提供技术:DIA定量蛋白组、靶向脂质代谢组学
技术路线:

研究结果
1ATG9A过表达破坏肝脏的自噬流
通过正常饮食(ND)和高脂高果糖饮食(HFD)建立正常小鼠和脂肪性肝病模型(图1A),尾静脉注射AVV-Atg9a-GFP过表达病毒,使Atg9a特异性的在肝脏中高表达(图1B-1E),导致肝脏中LC3-II水平升高,SQSTM1/p62积累,提示自噬降解存在障碍(图1F-1G)。利用mCherry-GFP双荧光报告系统发现,ATG9A可显著增加自噬体(黄),减少自溶酶体(红)(图1H-1I)。但不改变小鼠的肝脏重量和肝体比(图1J-1K)。
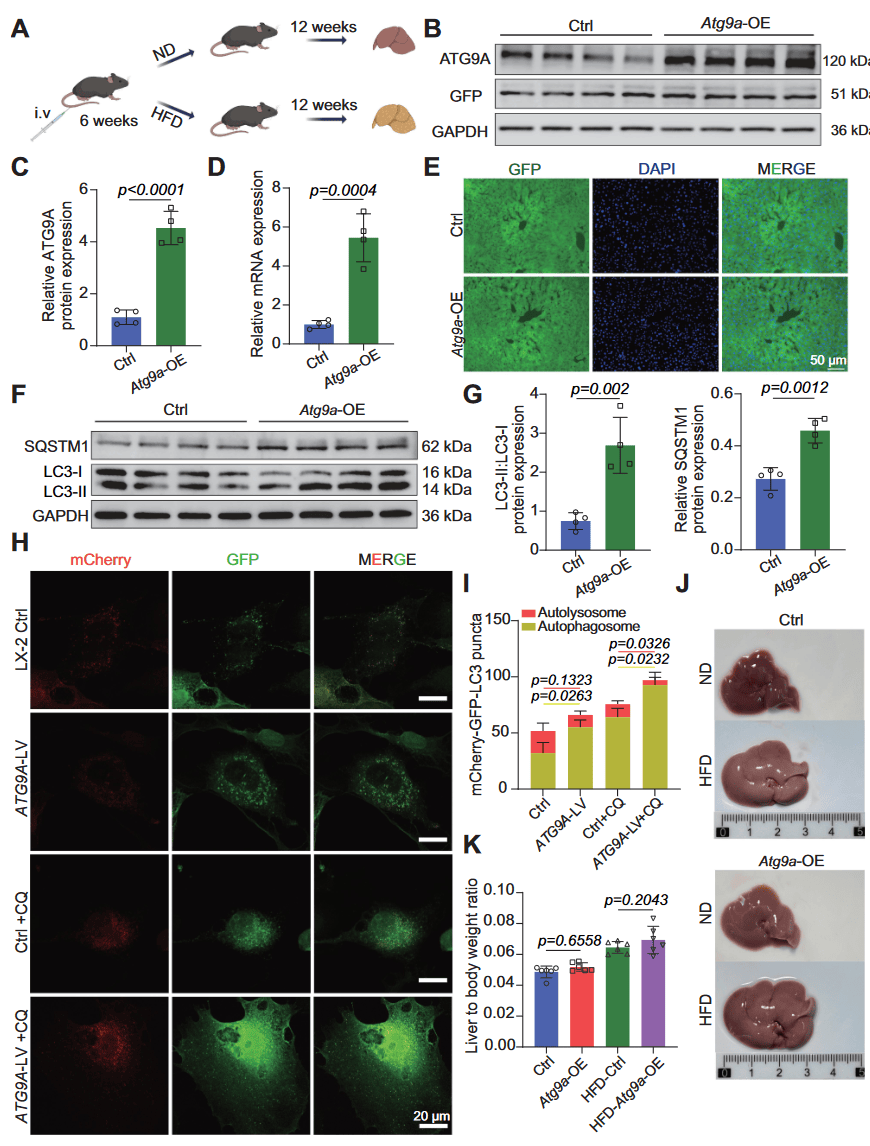
图1 ATG9A激活自噬流但损害自噬体降解
2ATG9A影响脂肪变性和肝纤维化
相比较ND小鼠,HFD Atg9a过表达小鼠出现更严重的炎症浸润和纤维化(图2A-2B),且血清中的中谷丙转氨酶(GPT)和谷草转氨酶1(GOT1)升高,证实肝脏发生损伤(图2C)。但油红染色和血清靶向脂质检测发现,HFD Atg9a过表达小鼠肝脏中的中性脂滴(LD)含量降低,甘油三酯(TAG)储存减少(图3A-3D)。血糖监测显示,HFD Atg9a过表达小鼠存在胰岛素抵抗(图3E)
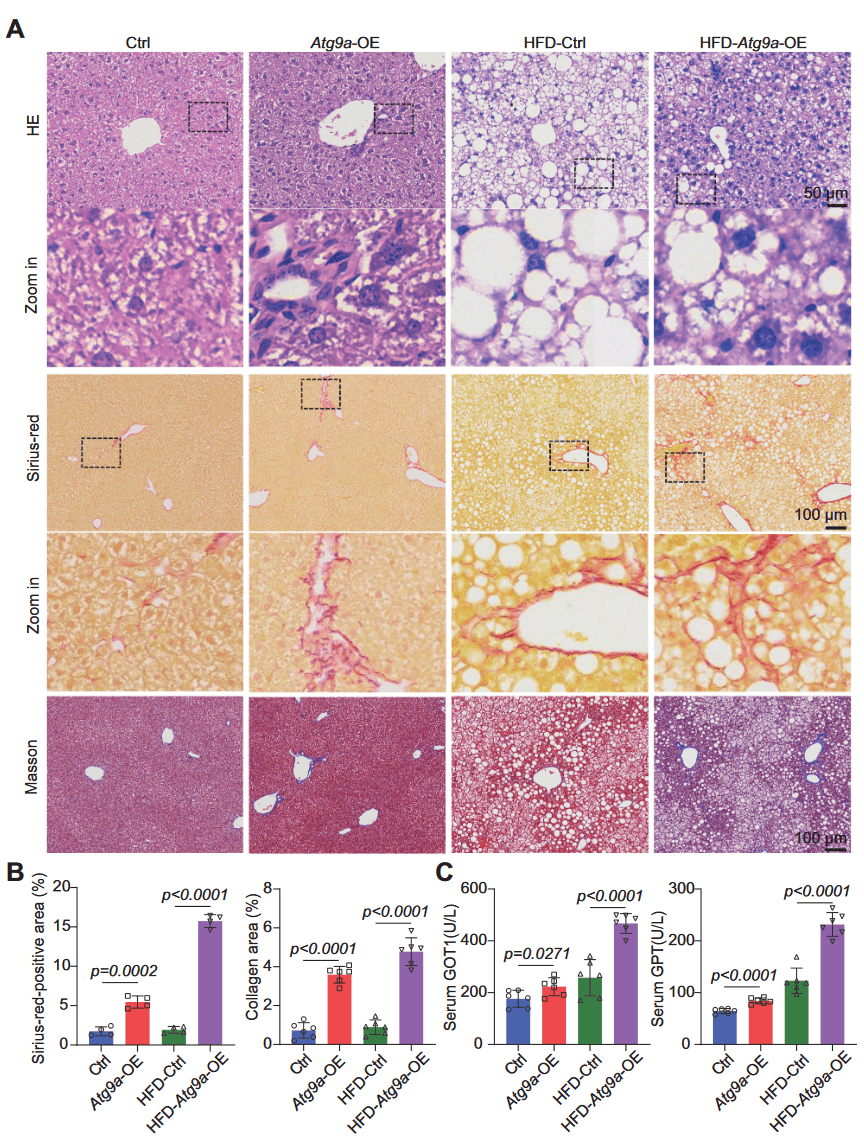
图2 ATG9A的高表达加剧了纤维化和炎症
3ATG9A过表达扰乱了甘油磷脂代谢
对Atg9a过表达小鼠和对照小鼠的肝脏样本进行蛋白质组检测,KEGG富集分析显示Atg9a过表达小鼠肝脏中线粒体自噬通路上调(图3F-3G)。靶向脂质代谢组学显示,Atg9a过表达肝脏中甘油磷脂代谢被影响,甘油磷脂类别减少(图4A-4B)。其中,甘油磷脂的降解酶PLA2G6表达上调,参与二酰基甘油(DAG)转化酶LPIN1表达下调(图4C-4G)。
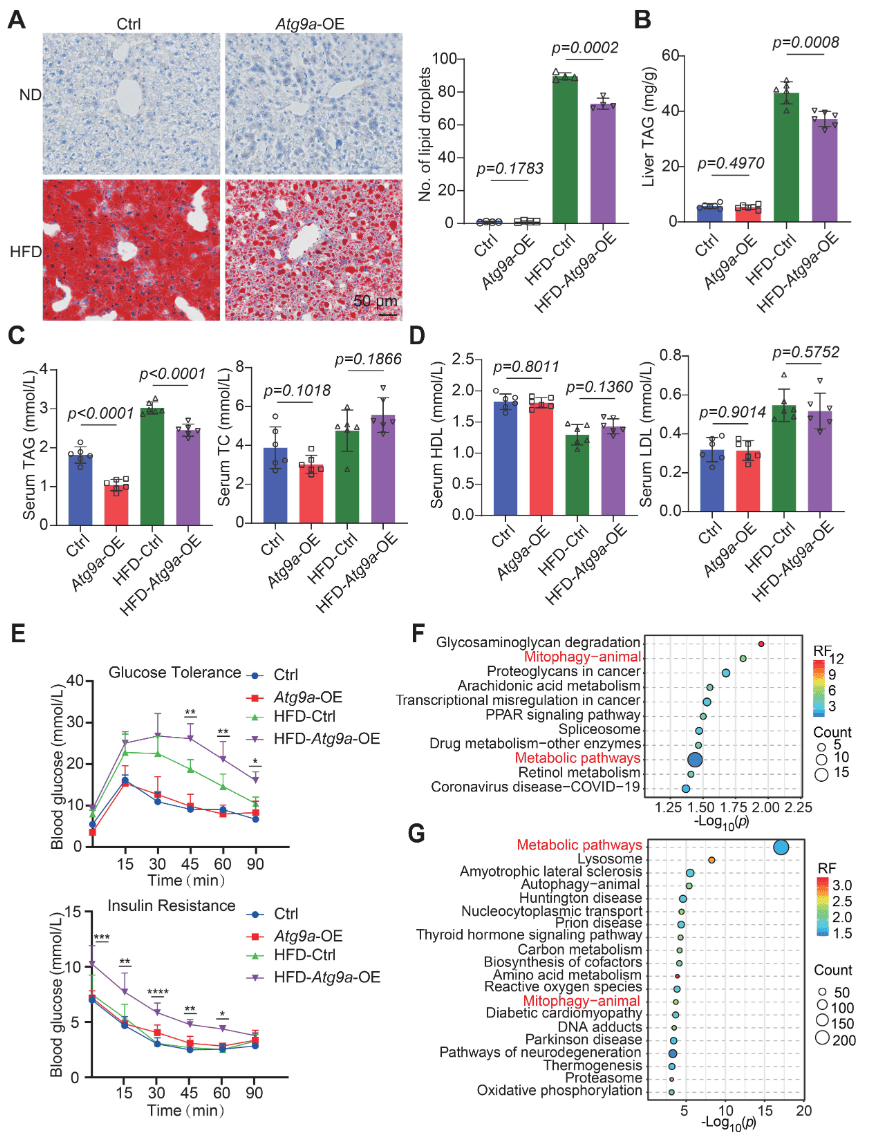
图3 ATG9A过表达对脂质稳态的破坏
4ATG9A与PLA2G6相互作用加速PC降解,激活炎症信号
通过免疫共沉淀(Co-IP)和液相色谱-质谱/质谱(LC-MS/MS)实验,检测到ATG9A与PLA2G6的结合(图4K-4M)。PLA2G6可催化磷脂酰胆碱(PC)水解,释放游离脂肪酸(FFA)、花生四烯酸(AA)和溶血磷脂酰胆碱(LPC)(图5A)。而Atg9a过表达肝脏中也观察到PC的显著减少和LPC的显著增加(图5B-5C)。
通过构建缺失ATG9A结合域的截短型PLA2G6变体(PLA2G6-trunc),发现其与ATG9A的互作消除,PC的降解程度降低(图5D-5F)。此外,PC水解产生的游离脂肪酸水平也增加(图5G),炎症介质PGE2和LTC4的水平升高(图5H-5I)。由此证明PLA2G6通过与ATG9A结合加速PC的降解,破坏脂质代谢,激活炎症信号。
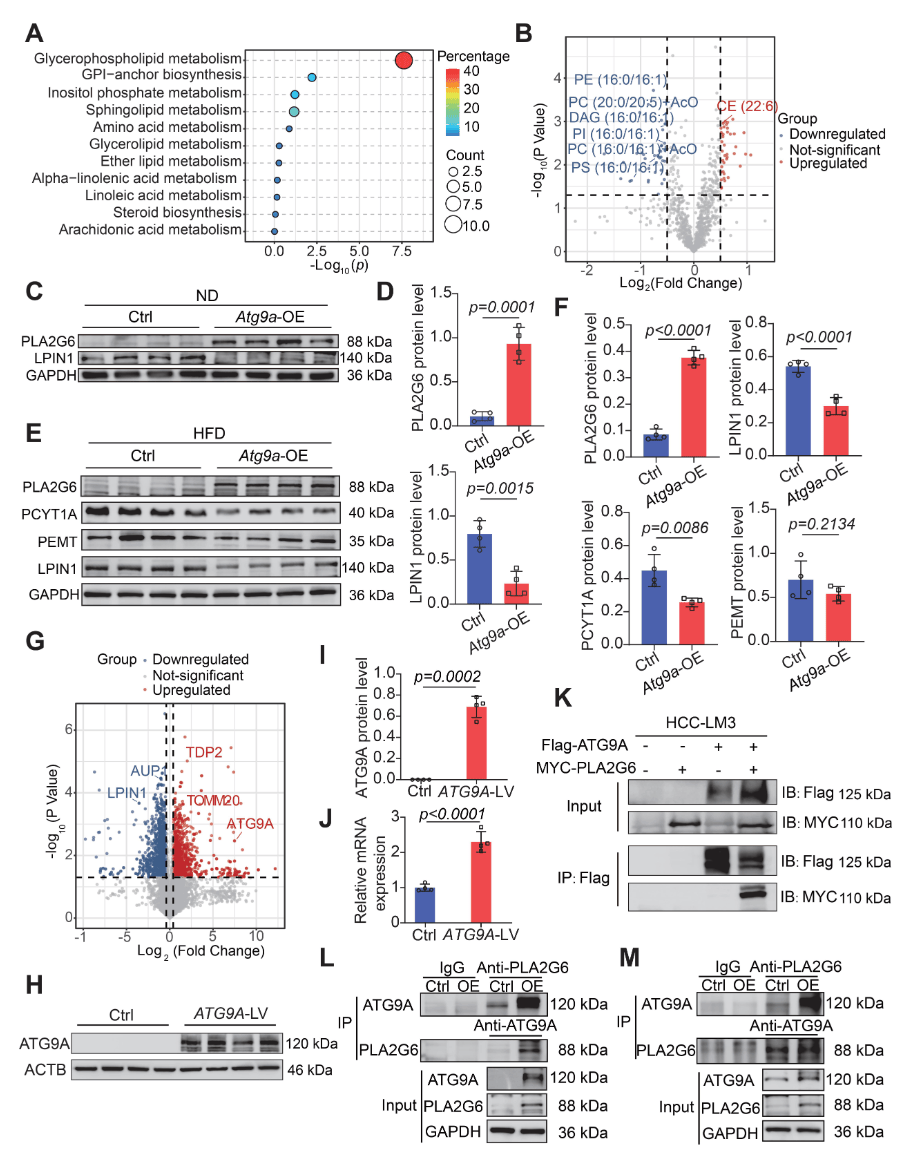
图4 ATG9A与PLA2G6结合并参与甘油磷脂代谢

图5上调ATG9A可加速PC分解代谢并激活炎症信号。
5ATG9A 过表达通过脂质过载导致线粒体功能障碍
已知游离脂肪酸可进入线粒体中进行氧化,通过超微结构分析发现,Atg9a过表达的肝细胞线粒体异常,包括嵴结构紊乱和碎片化(图6A),脂肪酸氧化(FAO)酶表达增强(图6B),ATP生成减少,呼吸能力受损(图6C-6F)。这些数据共同证实,ATG9A通过PLA2G6介导的水解作用,促使过量的游离脂肪酸流入β-氧化过程,超出了线粒体的代谢能力,引发细胞器功能障碍,直接诱导产生代偿性线粒体自噬。

图6 ATG9A过表达通过脂质过载导致线粒体功能障碍
6ATG9A-PLA2G6 轴以非自噬依赖的方式促进 HCC 进展
为了研究ATG9A在肝细胞癌(HCC)中的致癌作用,通过敲低和过表达在人HCC-LM3细胞中进行了功能分析。结果显示,ATG9A的表达与癌细胞的增殖、迁移和侵袭能力呈正相关(图7A-F)。通过在免疫缺陷小鼠中建立了细胞来源的异种移植(CDX)模型,证明ATG9A过表达的癌细胞表现出加速的生长动力学特征(图7G-7H),而这种加速迁移依赖于ATG9A与PLA2G6的结合(图7I-7J)。
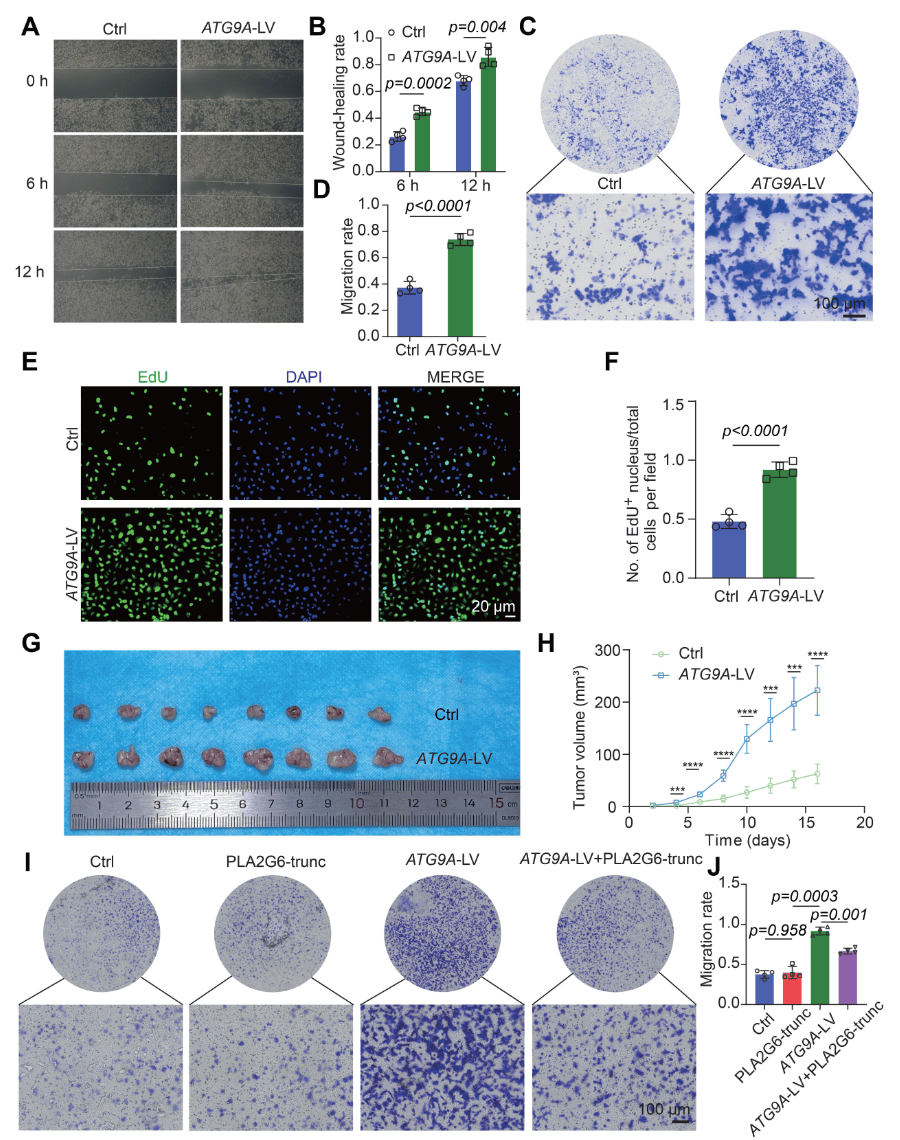
图7 肿瘤进展通过ATG9A-PLA2G6轴
为了进一步确认ATG9A对癌细胞线粒体自噬的影响,测定了自噬标志物,发现ATG9A不改变LC3和SQSTM1丰度(图8A-8B),对清除能力也没有显著改变(图8C-8D)。氯喹处理未能减少ATG9A驱动的细胞迁移(图8E-8H)。由此说明,自噬调节不是癌细胞增殖的主要机制。
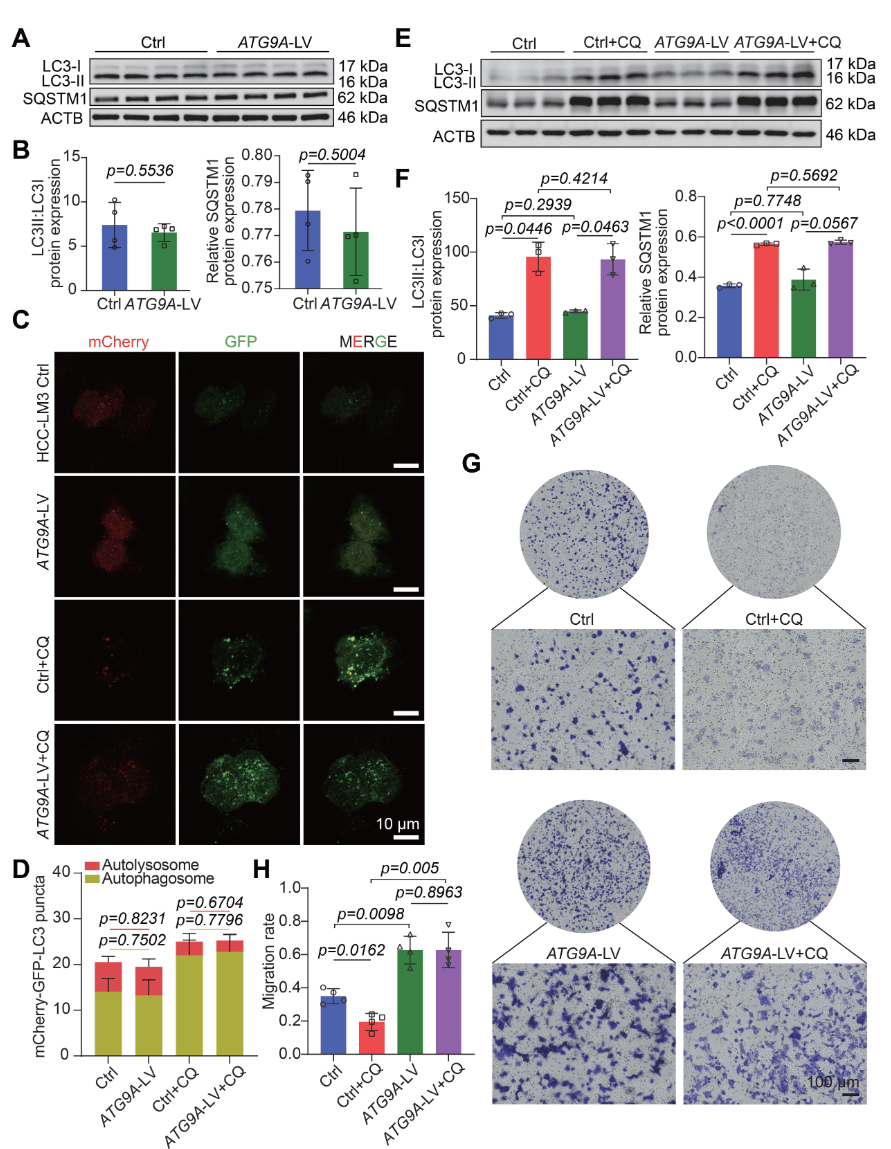
图8 ATG9A的非自噬功能促进了细胞迁移
ATG9A 过表达抑制 TAG 合成,阻碍脂滴生成
前文蛋白质组学分析显示,甘油三脂(TAG)生物合成的关键酶LPIN1表达降低。为了验证这一结果,在小鼠和细胞中检测了TAG关键合成酶——LPIN1和DGAT1。发现,ATG9A过表达导致LPIN1显著减少,DGAT1不变(图9A-9C),敲除从反面证实TAG合成能力受损(图9D-9F)。尼罗红染色显示,ATG9A过表达(ATG9A-LV)组的脂滴(LD)减少,抑制ATG9A组(Si-ATG9A)组的LD增加(图9G-9I)。由此证明,ATG9A可能是通过依赖PLA2G6的脂质代谢重编程促进肝癌的生长和转移,而非通过经典的自噬途径。
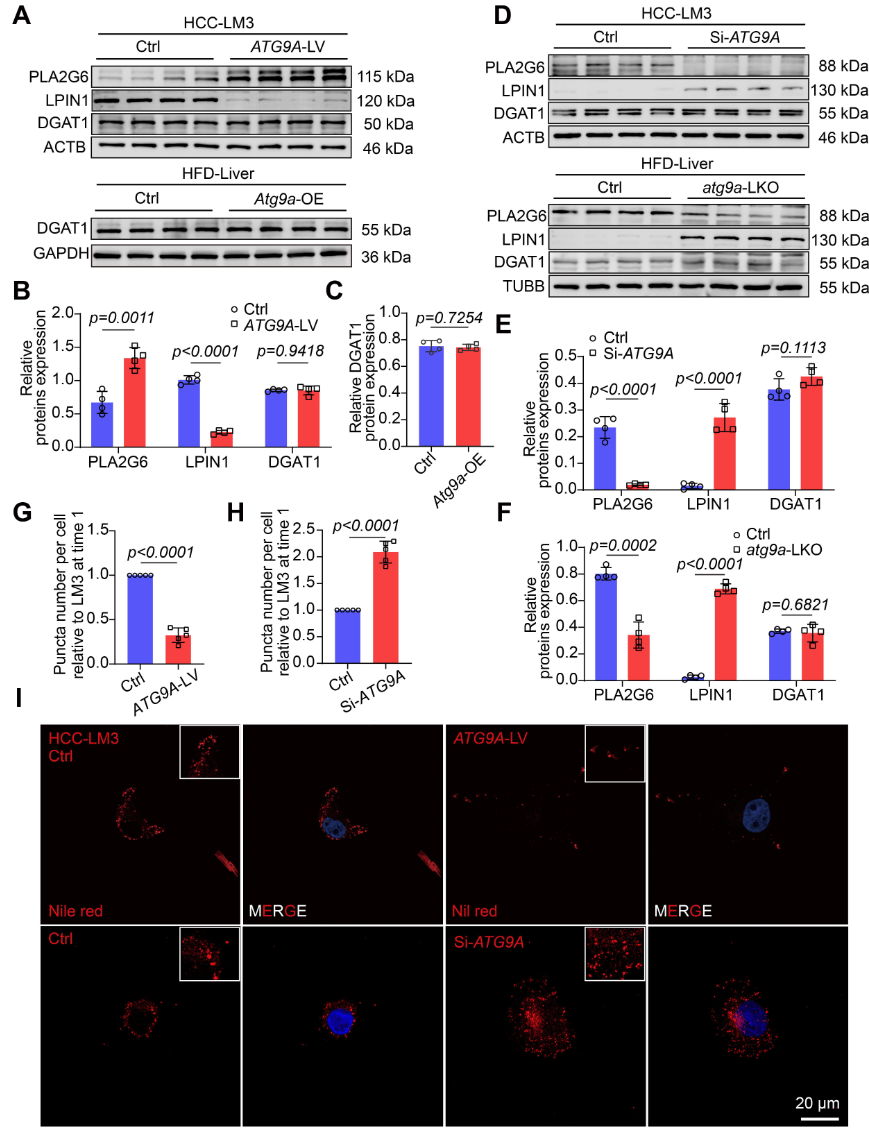
图9 ATG9A抑制TAG合成
文章小结
本研究通过小鼠模型、细胞实验及多组学分析证实,ATG9A在HCC组织中高表达,可与PLA2G6结合形成调控轴,加速PC降解,扰乱脂肪酸代谢并引发线粒体功能障碍。同时,以非自噬依赖方式加剧肝脏炎症与纤维化、促进HCC增殖和转移。该研究将ATG9A定义为一种双重代谢效应因子,可通过脂质重塑和细胞器应激驱动肝脏疾病进展,为代谢性肝病和肝细胞癌提供了一个治疗靶点。
拜谱小结
本研究揭示ATG9A与PLA2G6形成调控轴,通过重编程磷脂代谢引发线粒体功能障碍,以非自噬依赖方式驱动代谢性肝病(如MASLD)和肝细胞癌(HCC)进展,二者构成代谢性肝病及HCC的潜在治疗靶点。拜谱生物为该研究提供DIA 定量蛋白组学和靶向脂质代谢组学技术支持。拜谱生物可提供完善成熟的蛋白质组学、修饰蛋白质组学、代谢组学、转录组学等多组学产品技术服务体系,整合多组学数据进行深入挖掘分析,全面解析机制机理等,助力高分文章发表!
参考文献
Zhu Q, Gu Y, Gao Y, Zhao X, Zhang L. ATG9A-PLA2G6 axis reprograms phospholipid metabolism to drive metabolic liver disease and hepatocellular carcinoma. Autophagy. 2025 Dec 26:1-18. doi: 10.1080/15548627.2025.2601035.





关注公众号





关注小红书


