

400-820-8531




上期内容详细介绍了基于质谱的蛋白质实验流程的样本准备及LC-MS/MS分析(长篇综述|基于质谱的蛋白质组学简介(上)),本期将进一步介绍数据分析及蛋白质组学的前沿发展。
英文标题:An Introduction to Mass Spectrometry-Based Proteomics
期刊:Journal of proteome research
发表时间:2023年7月7日
 图1. 基于质谱的蛋白质组学概述(图源:Shuken, J Proteome Res, 2023)
图1. 基于质谱的蛋白质组学概述(图源:Shuken, J Proteome Res, 2023)
01.数据分析:多肽鉴定
单次LC-MS/MS运行产生的原始数据集是一个大的光谱集合,每个光谱都有保留时间、m/z值、强度和元数据。采用MaxQuant或Proteome Discoverer(Thermo Fisher Scientific)等软件包处理这些数据,生成肽和/或蛋白质列表,每个识别都有一个分数。大多数非靶向的自下而上的DDA蛋白质组学实验中使用的最常见的肽鉴定方法为数据库搜索(图2)。
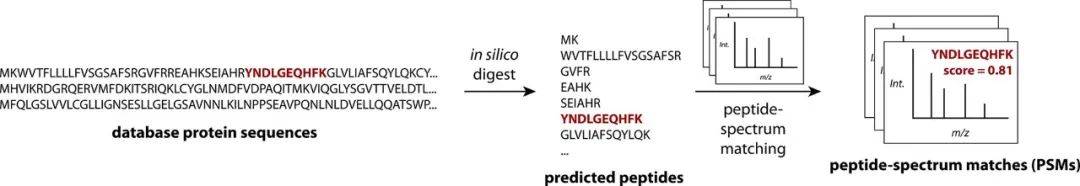 图2. 数据库搜索的简化概述(图源:Shuken, J Proteome Res, 2023)
图2. 数据库搜索的简化概述(图源:Shuken, J Proteome Res, 2023)
数据库搜索开始于用户将原始数据文件连同已知参考蛋白质组/数据库作为文本文件加载到软件中,通常采用FASTA格式。Uniprot.org拥有许多物种的参考蛋白质组,广泛用于人类和小鼠;某些物种可选用其它数据库。通过数据库搜索,数据处理软件预测通过酶切(最常见的是胰蛋白酶和LysC)从数据库中的蛋白质中产生的所有肽,并预测相应的电荷特异性肽离子(前体)的MS2光谱,作为肽的“指纹”。将这些预测肽及其预测光谱与实验光谱进行比较,得出肽谱匹配(psm)(图2)。在控制错误发现率(FDR)后,完成肽鉴定。
例如,小鼠己糖激酶1(Hexokinase-1)的氨基酸序列如图3所示,其中胰酶和LysC可能靶向的切割位点用橙色星号突出显示。下划线绿色区域由在小鼠脑组织LC-MS/MS分析中鉴定的肽序列组成。请注意,所有带下划线的区域都以精氨酸(R)或赖氨酸(K)残基结尾。图3中突出显示的肽(NILIDFTK)的MS2谱在图4中进行了注释。与预测片段离子相匹配的峰用蓝色“b”或红色“y”标记,并用整数表示片段的氨基酸长度。
 图3. 小鼠Hexokinase-1的氨基酸序列(图源:Shuken, J Proteome Res, 2023)
图3. 小鼠Hexokinase-1的氨基酸序列(图源:Shuken, J Proteome Res, 2023)
 图4. 片段m/z值、带注释的MS2谱,以及图3中突出显示的片段序列(NILIDFTK)
图4. 片段m/z值、带注释的MS2谱,以及图3中突出显示的片段序列(NILIDFTK)
1.1 数据库搜索算法示例:SEQUEST的简化描述
SEQUEST是1994年出版的首个全自动肽识别软件。图5演示了SEQUEST算法的简化概念。首先,对参考蛋白质组预测的肽进行过滤,以便只考虑与分离的断裂离子的m/z值相似的前体(即电荷状态特异性肽离子),然后生成理论光谱(图5,顶部)。通过将实验光谱与预测片段的m/z值进行粗略比较,理论光谱被快速过滤,匹配片段的数量、MS2强度和其它相关特征组合成一个分数,仅保留基于该分数的前500个理论光谱(图5,步骤1)。通过消除前体峰,将光谱划分为十个相等的区域,并将每个区域的强度归一化为相同的值来调整实验光谱(图5,步骤2),这一步使实验光谱更接近理论光谱,减少了构建理论光谱时忽略的可变碎片化效率的。通过将每个m/z值处的强度相乘并将乘积相加,计算实验光谱与每个理论光谱之间的相互关系(图5,步骤3)。
 图5. 简化了原SEQUEST数据库搜索算法的方案(图源:Shuken, J Proteome Res, 2023)
图5. 简化了原SEQUEST数据库搜索算法的方案(图源:Shuken, J Proteome Res, 2023)
1.2 肽鉴定中的FDR控制
我们如何决定哪些分数足够高来表示真实的ID?目前为止,最流行的方法是 Target-decoy搜索(图6)。该过程控制全局错误发现率(FDR),即被接受的PSM错误的平均比例。为了在控制FDR的同时最大限度地检测真肽,可以使用搜索分数和PSM的其它特征的线性组合(图6)。线性组合中使用的系数{a, b,…}被优化以最大限度地检测真肽,通常使用percolator使用的机器学习算法。在此优化完成后,所有超过所需FDR对应阈值的目标匹配(例如,1%)被保留。这个过滤数据集的FDR称为“全局”FDR。
 图6. Target-decoy搜索。PSM=肽谱匹配;FDR =错误发现率(图源:Shuken, J Proteome
图6. Target-decoy搜索。PSM=肽谱匹配;FDR =错误发现率(图源:Shuken, J Proteome
02.肽段定量
高效液相色谱在整个运行过程中连续地将多肽喷雾到质谱仪中。每个扫描周期一次,通常需要3 s或更少,光谱仪产生MS1光谱;因此,在高效液相色谱柱洗脱过程中,前体的MS1峰经常被观察到多次,这些MS1数据点一起形成色谱峰(图7A)。给定特定的肽,其色谱峰的特征,如其高度或曲线下面积(AUC),可用于测量肽的相对丰度,这被称为非标记定量(LFQ),该方法可用于不同样品中的数量比较。
为了提高定量重现性、样品通量和/或数据完整性,标记试剂可用于定量同一质谱中的多个生物样品。在这些方法中,除了不同的同位素导致肽或其片段的m/z值不同外,肽被标记为相同的原子群,同时保留化学性质,如保留时间、电离性和片段模式。在代谢标记方法中,如细胞培养氨基酸稳定同位素标记(SILAC),利用生物系统将含有重同位素的氨基酸标记整个蛋白质组。在样品制备开始时,将天然“轻”蛋白与重蛋白结合,然后与MS1光谱进行比较(图7B)。在等压法(即质量相等,因为不同的标签具有相同的总质量)标记方法中,如串联质量标签(TMT),肽段在消化后被标记,然后在LC-MS/MS前组合。高能CID(HCD)为每个生物样品释放不同的报告离子;通过MS2或MS3扫描测量报告离子强度,可以推断出相对丰度(图7C)。使用LFQ,每次运行分析一个生物样本;使用SILAC,每次运行通常分析两到三个生物样本;使用TMT,一次可以分析多达18种不同的生物样本。
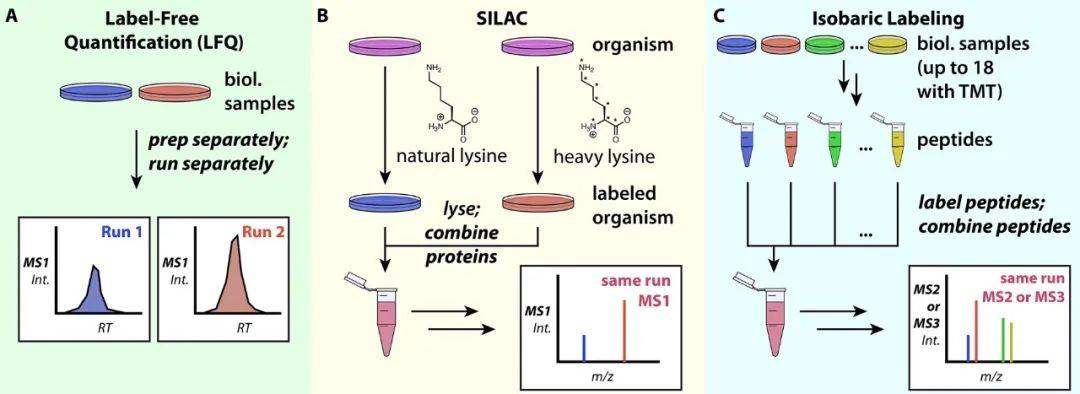 图7. 多肽定量方法。A.在MS1水平进行非标记定量;B. 细胞培养中氨基酸稳定同位素标记(SILAC);C. 等压标记
图7. 多肽定量方法。A.在MS1水平进行非标记定量;B. 细胞培养中氨基酸稳定同位素标记(SILAC);C. 等压标记
03.“蛋白质水平”:推断蛋白质的特性和丰度
3.1 蛋白质推断与分组
尽管一些使用自下而上蛋白质组学提到了“蛋白质”的身份和数量,但它们通常是鉴定和/或量化蛋白质群,其起源的蛋白质或基因可能是不明确的。对于这种歧义,最常见的解决方案是使用每个PSM作为样品中存在相应蛋白质的证据。如果一组蛋白质之间有相等的证据(即蛋白质序列与同一组鉴定的肽序列匹配,如图8中的蛋白质V和VI),则将这些蛋白质组合为一个蛋白质组(PG)。如果一个蛋白质的匹配肽是另一个蛋白质的子集,则该蛋白质通常被排除在报告之外,而缺乏该蛋白质特有肽但其肽不是子集的蛋白质称为可包含蛋白质。
 图8. 蛋白质推断和分组策略(图源:Shuken, J Proteome Res, 2023)
图8. 蛋白质推断和分组策略(图源:Shuken, J Proteome Res, 2023)
3.2 蛋白水平FDR控制
大多数执行蛋白质分组的流行软件包,如MaxQuant和Proteome Discoverer,也可以计算蛋白质水平鉴定FDR的估计,并将其重新调整到可接受的水平。有多种方法可以控制蛋白质水平的FDR。一种直接的策略是利用诱饵PGs,即完全由诱饵PSM组成的PG,它们被自动保留并受到与目标PSM相同的蛋白质推断过程。为每个目标和诱饵PG构建一个分数,允许以与PSM FDR类似的方式控制蛋白质水平FDR(图6)。
3.3蛋白组定量
多肽和蛋白质之间关系的多方面因素使这一过程复杂化。PG内的肽可能在它们匹配的蛋白质中彼此不同,这可能会影响它们的丰度(图8)。如果PG中的肽是该PG所独有的,这是有帮助的,允许对单个蛋白质进行量化。然而,复杂的是肽可能产生于含有翻译后修饰的蛋白质,其丰度影响肽的强度,此外,由于DDA固有的随机性,一些肽可能在一个样品中观察到而在另一个样品中观察不到。流行的数据处理软件包为用户提供了解决这些问题的选项。大多数软件(包括MaxQuant)只允许对PG-unique肽进行定量。一些(如Spectronaut ,Biognosys)允许更多的规格,如限制定量到蛋白质特异性肽,或选择是否使用肽强度的中位数、和、算术平均值或几何平均值来计算PG数量。对于LFQ,值得注意的是MaxQuant中实现的流行的MaxLFQ算法。当在不同的样本中识别出不同的前体集时,MaxLFQ通过采用可用的两两比较和使用中位数比率来比较PG来处理缺失值。等压标记和数据独立采集(DIA)是减少或消除缺失值的其它方法。
3.4 统计分析和生物学解释
一旦计算出PG的相对数量,根据实验的生物学特性,可以使用各种工具对结果进行统计分析和解释。Perseus和MSStats是设计用于蛋白质组数量处理和统计分析的流行软件包。数据也可以使用编程语言(如python或R)手动分析。在一般比较蛋白质组学实验中,包括从列表中去除污染物或诱饵,使用对数使数量近似正态分布,将数据归一化以纠正运行间的技术变异性,使用统计检验(如t检验/方差分析/线性回归)检验变化,并对结果p值进行多假设检验。
统计分析完成后,结果可以进行生物学解释。Gene Ontology是研究在实验中发生改变的生物途径的一个流行数据库。STRING提供了有关感兴趣的蛋白质之间已知关系或相互作用的信息。相关分析,如加权基因相关网络分析(WGCNA),根据实验中蛋白质丰度变化的相似程度构建蛋白质网络,这可以进一步了解实验中蛋白质之间的关系。通过综合解释这些分析的结果,生物学假设可以得到支持或反驳,并且可以产生新的假设。
04.拜谱生物总结:质谱工作流程
总的来说,非标记的自下而上的工作流程如下:
(1)分离组织/植物、溶解细胞;用尿素和/或去垢剂使蛋白质变性。
(2)用DTT或TCEP等方法减少二硫化物;用IAA烷基化半胱氨酸。
(3)用冷丙酮、氯仿-甲醇沉淀法或磁珠沉淀法部分纯化蛋白质(有些工作流程不使用有害的去垢剂如SDS,可跳过这一步)。
(4)用胰蛋白酶或LysC组合蛋白酶消化蛋白质。
(5)用C18包被的过滤吸管头离心清洗/脱盐多肽;用真空离心机等蒸发溶剂。
(6)将肽重悬于LC-MS兼容的溶剂中,例如0.1%甲酸水溶液;采用相应的仪器方法进行LC-MS/MS分析;在运行完成后检索原始数据。
(7)使用合适的软件包处理原始数据,如MaxQuant或Proteome Discoverer,或软件包的组合,执行数据库搜索、FDR控制、相对定量和蛋白质推断。
(8)使用编程语言(如python或R)或软件包(如Perseus或MSStats)执行统计分析。
(9)根据实验的生物学原理来解释结果;这可以通过路径或网络级分析资源(如Gene Ontology、STRING或WGCNA)来辅助。
05
前沿主题
除了DDA进行的非靶向自下而上的无标记蛋白质组,还有更多的基于质谱的方法来研究蛋白质组(图9)。仪器和算法均在与时俱进的更新,完整蛋白质、自上而下和天然质谱法也用于分析低复杂性的样本;蛋白的翻译后修饰因其至关重要的调控作用拥有广阔的研究天地。结构蛋白质组学、化学蛋白质组学和接近标记等技术使用化学、酶或热技术来探测蛋白质,为研究蛋白质结构、化学、定位、蛋白质-配体相互作用或蛋白质-蛋白质相互作用开辟了大量的机会。相比DDA,数据独立采集(DIA)是一种越来越受欢迎的采集方法,最流行的DIA方法是基于SWATH-MS,其中整个MS1范围内的所有m/z值在每个扫描周期内都包含在碎片中,这大大提高了数据完整性,增加了蛋白质组学深度。在靶向蛋白质组学中,一种特定的蛋白质或一组蛋白质被作为分析的目标,其目标是确保每次方法运行时都能检测到目标蛋白质(数据完整性高),最大限度地提高灵敏度和动态范围,以及定量准确性和精密度。
对于大规模的蛋白质鉴定和定量,质谱法是目前最流行的方法,在未来,质谱与其它非质谱技术的结合使用可以更深入地了解整个生物学中的蛋白质组。
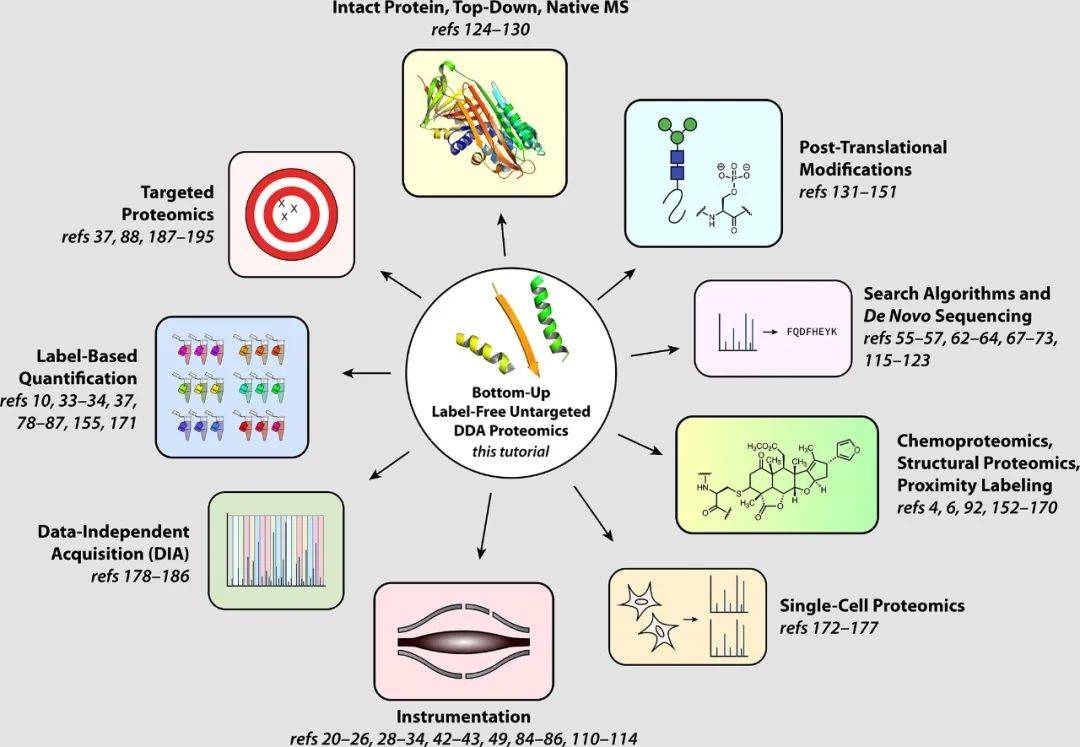 图9. 基于质谱的蛋白质组学的子领域(图源:Shuken, J Proteome Res, 2023)
图9. 基于质谱的蛋白质组学的子领域(图源:Shuken, J Proteome Res, 2023)
参考文献:
Shuken SR. An Introduction to Mass Spectrometry-Based Proteomics. J Proteome Res. 2023; 22(7):2151-2171. doi: 10.1021/acs.jproteome.2c00838.





关注公众号





关注小红书


