

400-820-8531




质谱方法在蛋白质组学研究中起到无与伦比的作用,但基于质谱的蛋白质组学基础复杂且跨越多个科研领域,由此蛋白质组学有很高的进入门槛。本综述旨在为相对简单的定量蛋白质组学实验的技术细节提供一个易于理解的图解指南。本综述进行了基于质谱的蛋白质组学实验概述,从样品准备到蛋白质组数据分析,并解释了数据是如何获得、处理和分析的。最后,我们对蛋白质组学的未来进行了简短讨论,探讨了可能与质谱相辅相成的下一代蛋白质测序技术,为蛋白质组学创造出更为丰富的未来。
标题:An Introduction to Mass Spectrometry-Based Proteomics
期刊:Journal of proteome research
发表时间:2023年7月7日
 图1. 基于质谱的蛋白质组学概述(图源:Shuken, J Proteome Res, 2023)
图1. 基于质谱的蛋白质组学概述(图源:Shuken, J Proteome Res, 2023)
一、简介
以质谱为基础的蛋白质组学可以解决关于蛋白质的一系列问题,如蛋白质的序列、表达丰度、亚细胞定位、蛋白功能、三维结构、化学反应性及蛋白质-蛋白质相互作用等。这项技术的核心仪器便是质谱仪,它可以检测不同复杂程度的样品中蛋白质的质量电荷比(m/z)和信号强度。
基于质谱的蛋白质组学最常见的技术之一是使用数据依赖式采集(data-dependent acquisition,DDA)对样品(细胞、组织、体液或者植物、真菌等材料)进行非靶向自下而上(bottom-up)的定量蛋白质组学实验。短语“自下而上”是指从肽分析中推断蛋白质的信息,这类型研究的方法是从样品中提取蛋白质,酶解为肽段,然后采用质谱分析多肽(通常7-30个氨基酸较为适宜)(图2)。当以DDA模式获取质谱数据时,检测到肽,然后立即逐个选择进行片段化,以便在下游数据分析中分配其序列。与完整的蛋白质相比,多肽有以下几个优点:较小的大小分散分布、更适合反相高效液相色谱(HPLC)分离,并产生更容易解释的片段光谱。
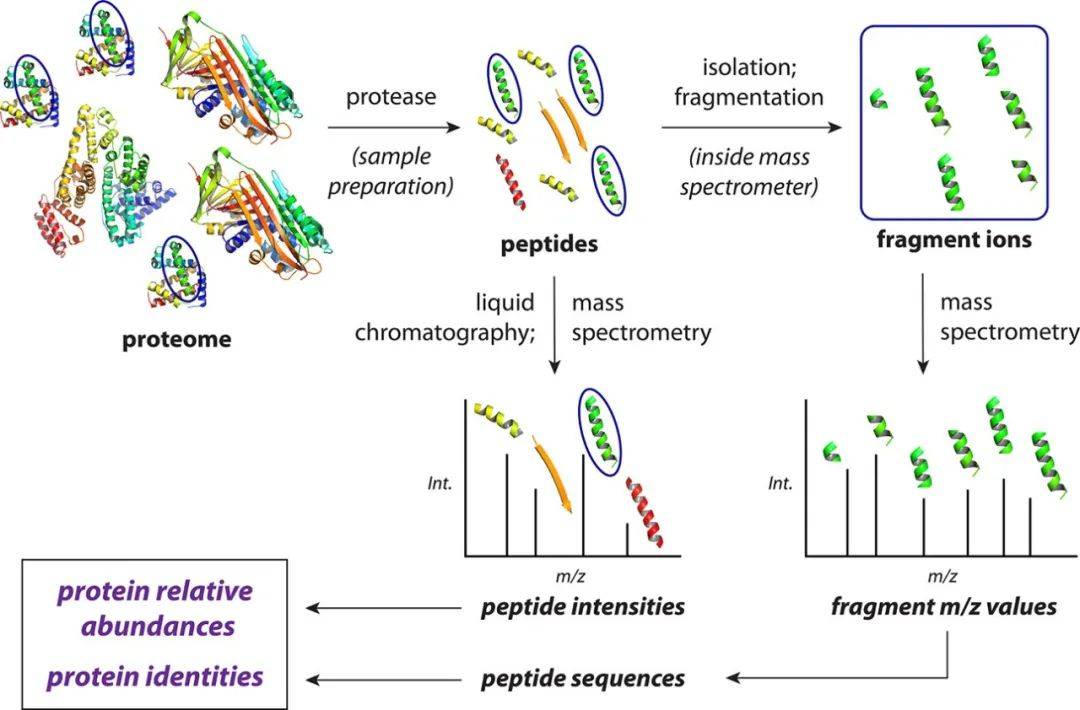 图2. 典型的非靶向自下而上质谱蛋白质组实验的简化概念概述(图源:Shuken, J Proteome Res, 202
图2. 典型的非靶向自下而上质谱蛋白质组实验的简化概念概述(图源:Shuken, J Proteome Res, 202
二、实验流程
2.1 样本制备
每个蛋白质组实验都从样品制备开始,自下而上蛋白质组学的通用样品制备工作流程如图3所示。整个过程包括蛋白质的提取、变性、使用二硫苏糖醇(DTT)或磷酸三氯乙基酯(TCEP)还原二硫键、使用碘乙酰胺(IAA)将巯基烷基化,以及随后被LysC和胰蛋白酶“消化”。消化后的肽段需经C18包被的固相进行脱盐,最后将肽重悬于液相色谱-串联质谱(LC-MS/MS)分析兼容的水缓冲液中,此时肽可以进行LC-MS/MS分析(图3)。
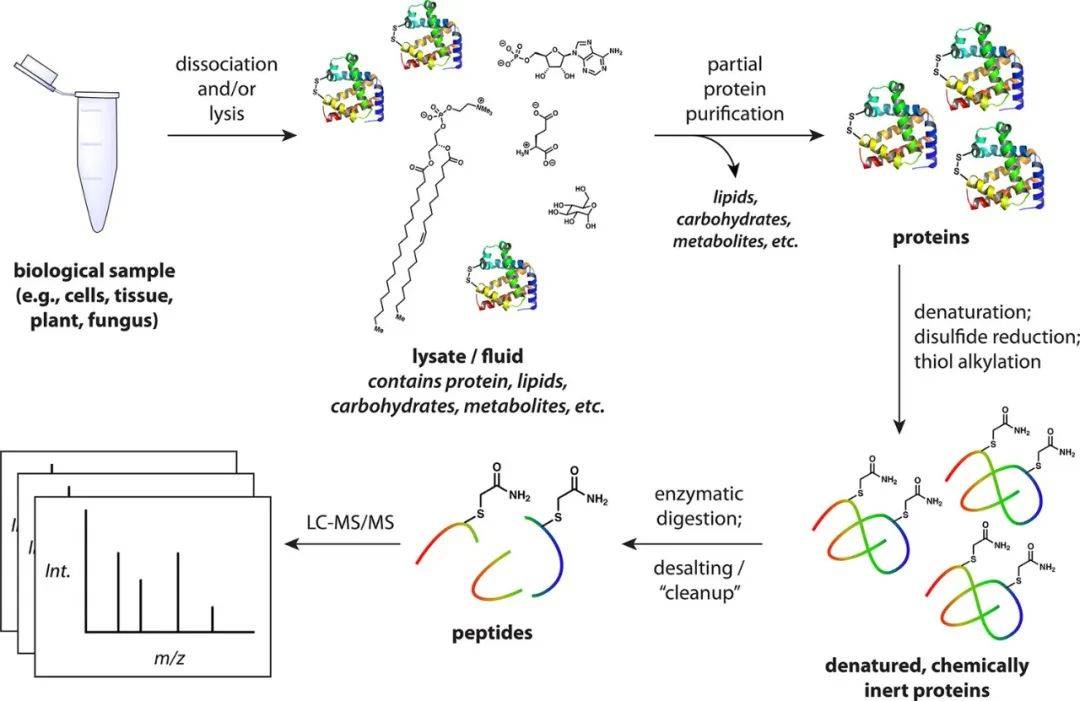 图 3. 基于自下而上质谱的蛋白质组学的通用样品制备工作流程(图源:Shuken, J Proteome Res, 20
图 3. 基于自下而上质谱的蛋白质组学的通用样品制备工作流程(图源:Shuken, J Proteome Res, 20
2.2 LC-MS/MS分析
LC-MS/MS运行开始后的第一步是样品提取和上样到HPLC柱上。根据仪器的不同,这个过程可以以多种不同的方式发生;图4展示了一个简单的示例。在使用高效液相色谱的非靶向蛋白质组学方法中,通常涉及通过施加负压(拉)然后施加正压(推)将特定数量的样品加载到高效液相色谱柱上。一旦肽被装载到色谱柱上,就会同时发生四件事:液相色谱、气相肽离子的形成(电离)、肽的质谱分析(MS1)和片段的质谱分析(MS2)。
 图4. LC-MS/MS中高效液相色谱法的简化说明(图源:Shuken, J Proteome Res, 2023)
图4. LC-MS/MS中高效液相色谱法的简化说明(图源:Shuken, J Proteome Res, 2023)
2.2.1 HPLC
样品通过使用预编程溶剂梯度的高效液相色谱柱洗脱,即在整个运行过程中改变溶剂混合物的成分,其长度通常为30-180分钟。在蛋白质组学中,高效液相色谱几乎总是以反相模式进行,这意味着柱中填充了疏水固定相(通常是二氧化硅包被长度为18个碳的线性烃链,称为C18)。溶剂(流动相)通常是两种溶液的混合物(水溶液如0.1%甲酸和有机溶液如乙腈),每种溶液都由自己的梯度泵泵送。肽根据其疏水性被部分分离,在梯度过程中在不同时间(保留时间,RTs)洗脱。最佳梯度取决于几个因素,包括色谱柱、样品复杂性、仪器和实验目标。图5显示了一个使用HPLC梯度进行肽分离的例子,色谱图中的每个峰代表一组不同的离子。
 图5. LC-MS/MS运行的典型色谱图,样品为胰蛋白酶和LysC消化的小鼠脑蛋白(图源:Shuken, J Prote
图5. LC-MS/MS运行的典型色谱图,样品为胰蛋白酶和LysC消化的小鼠脑蛋白(图源:Shuken, J Prote
2.2.2 肽电离
随着梯度的进行,部分分离的肽被连续地喷射到光谱仪中。在施加电压的影响下,从HPLC柱顶端喷射出含有多肽的带电液滴;带电荷的肽被脱溶(进入气相),进入光谱仪,并在光谱仪内的电磁场推动下向前推进(图6)。给定的肽在电离时通常可以采用不同的电荷状态;每个电荷状态通常对应于质子化状态(有多少质子与肽结合)。不同的电荷状态导致不同的m/z值:例如,z = 1的电离肽将具有m/z = [m + H],其中m是中性肽的质量,H是质子的质量,而z = 2的相同肽将具有m/z = [m + 2H]/2。肽的不同电荷状态被特异性分离,称为肽的不同“前体”。
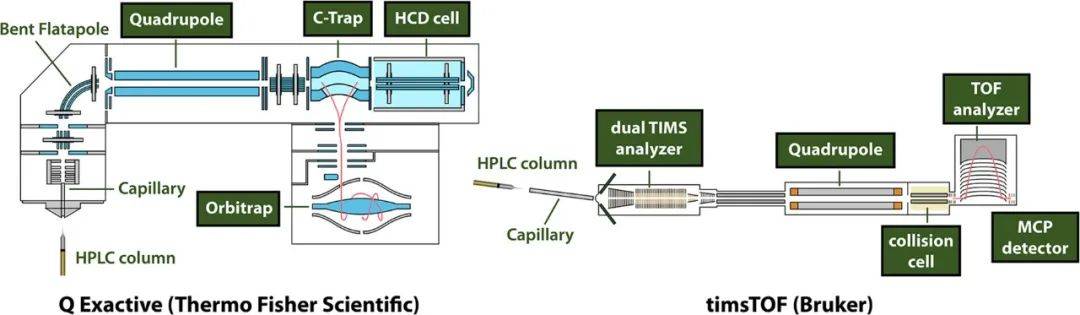 图6. 两种流行的蛋白质组学质谱仪示意图(图源:Shuken, J Proteome Res, 2023)
图6. 两种流行的蛋白质组学质谱仪示意图(图源:Shuken, J Proteome Res, 2023)
2.2.3 肽质谱扫描(MS1)
光谱仪反复快速获取光谱以检测电离肽。这些频谱采集事件被称为“MS1扫描”。任何质谱的获取都需要一个质量分析仪,它根据它们的质量电荷比(m/z)值来解析离子,以及一个测量电信号的探测器。两个流行的质量分析仪和检测器是飞行时间分析仪和轨道阱(图7)。
Q Exactive(Thermo Fisher Scientific)是相对简单的轨道阱质谱仪,MS1扫描的范围在400 ~ 1600 Th之间(Th为m/z的单位)。多肽进入C -Trap中,在吸收动能的氮(N2)分子的帮助下,经过一定的离子积累时间(“注入时间”),C -Trap同时并立即将其离子送入轨道阱。一旦进入轨道阱,离子围绕中心主轴进动,并以与它们的m/z值成比例的频率沿主轴振荡。当它们移动时,会产生波动电流,通过傅里叶变换处理得出m/z值和强度。这种质量分析方法具有高m/z分辨率和精度。测量完成后,生成如图7A所示的MS1频谱。
 图7. MS1和MS2光谱的例子。A. MS1频谱;B. MS2频谱(图源:Shuken, J Proteome Res
图7. MS1和MS2光谱的例子。A. MS1频谱;B. MS2频谱(图源:Shuken, J Proteome Res
另外一种质谱仪timsTOF(Bruker)和Q Exactive之间的一个主要区别是添加了捕获离子迁移谱(TIMS)(图6)。当肽进入timsTOF时,它们在双TIMS分析仪的第一个区域积累25 - 200 ms,然后在TIMS分析仪的第二部分被气流定位在不同的位置,而气流的方向是纵向梯度电场。离子在双TIMS分析仪第二部分的位置与其通过气体的能力有关(“迁移率”,与碰撞截面成反比)。然后在25 - 200 ms的过程中逐渐从双TIMS分析仪中“洗脱”,从而对不同的前体进行额外的分离。由于TOF的质量分析和MCP的检测需要很短的时间(每次扫描约0.1 ms),因此在双TIMS分析仪排空期间获得了许多MS1光谱,每次MS1采集都可能在下一个MS1之前触发MS2光谱,就像Q Exactive一样。
2.2.4 碎片MS扫描(MS2)
MS1采集测量电离肽的m/z值和强度,但缺乏确定肽身份/序列所需的信息。虽然m/z值通常具有很高的准确性,但在m/z误差允许范围内存在太多可能的肽,无法仅根据m/z来识别肽。为了获得鉴定所需的信息,将多肽片段化(通常在肽键处),用质谱仪测量片段,所得光谱称为“MS2光谱”。
在每次MS1扫描后,光谱仪在运行时可能会选择在MS1光谱中观察到的一些离子进行重新积累和碎片化。虽然存在多种前体选择策略,但通常选择光谱中强度最高的前体。为了避免重复选择先前碎片化的肽,最近碎片化的m/z值被排除在选择之外。
为了确定质谱峰是否可能代表肽而不是污染离子,光谱仪寻找邻近的重同位素峰,如13C和15N。一个碳都是12C的离子的质量(m)比含有一个13C的相同离子的质量(m)低1.003 Da,含13C离子对应的MS峰的m/z值大约高1/z,如z = 1时高1.0 Th, z = 2时高0.5 Th,z = 3时高0.33 Th,依此类推。因此,给定任何观察到的峰,如果在这些m/z距离上发现邻近的峰,则可以推定离子的电荷。这些峰的相对强度也可以估计出离子中有多少C和N原子,因此离子是肽的可能性有多大(图8);在给定的m/z下,肽的典型同位素峰强度比不同于洗涤剂或碳水化合物等其它分子。由于许多非肽污染离子具有+1电荷,并且z = +1的肽片段通常信息较少,通常选择带+2或更大电荷的离子进行碎裂。
 图8. 模拟质量增加阳离子的MS1谱(图源:Shuken, J Proteome Res, 2023)
图8. 模拟质量增加阳离子的MS1谱(图源:Shuken, J Proteome Res, 2023)
使用碰撞诱导解离(CID)将每个选定的前体碎片化,并获得碎片的MS2谱,这些MS2光谱被用来鉴定肽。离子被加速进入高能CID(HCD)池或碰撞池,在那里它们与气体分子(通常是N2)碰撞。碰撞诱导质子在解离之前从肽的某处转移到肽的一个主酰胺键上。CID倾向于优先切割羰基碳和酰胺氮之间的C - N键(即肽键),产生m/z值可预测的片段,称为b离子和y离子,b离子包含肽的C端,y离子包含肽的N端(图9),产生的碎片进行类似于MS1的质量分析和检测过程(图7B)。
 图9. 基于移动质子模型的碰撞诱导解离(CID)破碎事件的可能机制(图源:Shuken, J Proteome Res,
图9. 基于移动质子模型的碰撞诱导解离(CID)破碎事件的可能机制(图源:Shuken, J Proteome Res,
2.2.5 小结
整个梯度过程中(通常为30 - 180分钟),光谱仪不断积累气相离子。MS1离子被评估为肽样性质进行分析,每个假定的肽被重新积累和碎片化(MS2),然后循环往复。在Q Exactive中,如果一次MS1扫描耗时50 ms,并触发10次MS2扫描,每次扫描耗时200 ms,那么这个周期将在大约2 s内完成。在timsTOF中,一个典型的周期需要25 ~ 200 ms。这个周期在整个梯度过程中重复数百到数千次,产生数千个光谱。这种采集模式被称为数据依赖采集(DDA),因为获取MS2频谱的决定取决于MS1数据;另一种方法是数据独立采集(DIA)。如上所述,不充分电离的肽不会被检测到,但肽也必须很好地碎片化,即产生足够有信息的碎片以供识别(图9)。
三、拜谱生物小结
本期内容详细介绍了基于质谱的蛋白质实验流程的样本准备及LC-MS/MS分析,下一篇将介绍数据分析及蛋白质组学前沿发展,敬请期待!
参考文献:
Shuken SR. An Introduction to Mass Spectrometry-Based Proteomics. J Proteome Res. 2023; 22(7):2151-2171. doi: 10.1021/acs.jproteome.2c00838.





关注公众号





关注小红书


